
Experiment-Based Kinetic Monte Carlo Simulations: CO Oxidation over RuO2(110)
Published:
Abstract
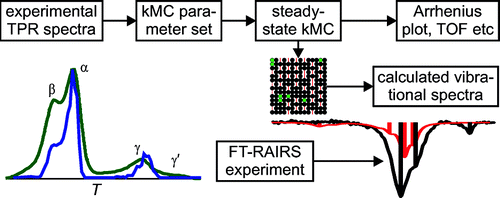
Kinetic Monte Carlo (kMC) simulations of the CO oxidation over RuO2(110) have been performed for a variety of different reaction conditions ranging from 10–7 to 10 mbar and temperatures from 300 to 600 K. The kMC simulations are based on reaction rates of elementary steps including diffusion, adsorption/desorption, and recombination of surface CO and O. The activation barriers for these elementary processes are mostly taken from temperature programmed reaction and desorption experiments of well-defined coadsorbate layers on RuO2(110) under ultrahigh vacuum conditions. We show that the experimental kinetic reaction data under steady state reaction conditions both in the 10–7 mbar range and in a batch reactor up to 10 mbar are reconciled within this experiment-based kMC approach. Experimental in situ reflection absorption infrared (RAIR) spectra in the frequency range of the CO stretch vibration depend sensitively on both the adsorption site and the local environment of the CO molecules, encoding thus the surface distribution of CO and O during the reaction experiment. Simulated RAIR spectra of kMC-determined snapshots of the surface configuration of reactants under reaction conditions reproduce well the experimental ones. RAIR spectroscopy provides thus a clear-cut criterion for assessing the quality of kMC simulations in the CO oxidation on RuO2(110).
Links
A. Farkas, F. Hess, H. Over. J. Phys. Chem. C. 116 (2012) 581-591. 10.1021/jp204703p