
Evidence of a Tetrahedrally Coordinated RuO4 Surface Complex on RuO2(100): Density Functional Theory and Beyond
Published:
Abstract
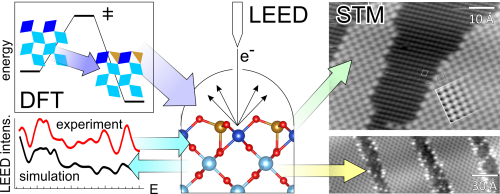
RuO2(100), an efficient low-temperature oxidation model catalyst, is able to undergo a c(2 × 2) reconstruction with a so far unknown surface structure that is accompanied by the loss of its catalytic activity. Here, the surface structure of the c(2 × 2) reconstruction of RuO2(100) is predicted by ab initio molecular dynamics simulations and shown to be consistent with available surface science experiments from the literature, including scanning tunneling microscopy (STM) images, low-energy electron diffraction (LEED) intensity versus voltage experiments, and thermal desorption experiments of O2. Mechanistically, the c(2 × 2) reconstruction starts from a simple c(2 × 2) overlayer structure of on-top O on RuO2(100), where the remaining undercoordinated surface Ru centers shift laterally toward the on-top O position, thereby breaking two Ru–O back bonds to lattice oxygen and forming a new Ru–O bond with on-top O. This leads to the formation of a tetrahedrally coordinated surface species Ru4f, a process that is activated by 0.95 eV and that results in an energy stabilization of 0.66 eV. The c(2 × 2) surface structure can readily explain the lack of activity in catalytic CO oxidation. The CO molecule is much more weakly bound on RuO2(100)-c(2 × 2) reconstruction than on the RuO2(110) or the RuO2(100)-(1 × 1) surface, and in addition, the Ru4f centers are too far apart to allow for O2 activation. The identified Ru4f surface species is expected to play a prevailing role as precursor species in the missing thermal and electrochemical stability of RuO2 under oxidizing reaction conditions by forming volatile RuO4.
Links
F. Hess, S. Rohrlack, M. Knapp, and H. Over. J. Phys. Chem. C 126 (2022) 946-956. 10.1021/acs.jpcc.1c08787