
Efficient implementation of Cluster Expansion models in surface Kinetic Monte Carlo simulations with lateral interactions: Subtraction Schemes, Supersites and the Supercluster Contraction
Published:
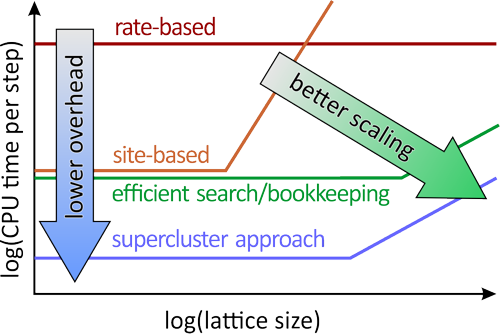
Highlights
- Scaling of KMC algorithms based on the Variable Step Size Method (VSSM) is discussed
- Impact of including lateral interactions on KMC performance is analyzed
- The supercluster approach to speed up computation of the cluster expansion is introduced
- Further improvements to VSSM are proposed and the scaling for a combination of all algorithms is demonstrated
Abstract
While lateral interaction models for reactions at surfaces have steadily gained popularity and grown in terms of complexity, their use in chemical kinetics has been impeded by the low performance of current Kinetic Monte Carlo (KMC) algorithms. The origins of the additional computational cost in KMC simulations with lateral interactions are traced back to the more elaborate Cluster Expansion Hamiltonian, the more extensive rate updating, and to the impracticality of rate-catalog-based algorithms for interacting adsorbate systems. Favoring instead site-based algorithms, we propose three ways to reduce the cost of KMC simulations: 1. Represent the lattice energy by a smaller Supercluster Hamiltonian without loss of accuracy, 2. employing Subtraction Schemes for updating key quantities in the simulation that undergo only small, local changes during a reaction event, and 3. applying efficient search algorithms from a set of established methods (Supersite Approach). The resulting algorithm is fixed-cost with respect to the number of lattice sites for practical lattice sizes and scales with the square of the range of lateral interactions. The overall added cost of including a complex lateral interaction model amounts to less than a factor 3. Practical issues in implementation due to finite numerical accuracy are discussed in detail, and further suggestions for treating long-range lateral interactions are made. We conclude that, while KMC simulations with complex lateral interaction models are challenging, these challenges can be overcome by modifying the established Variable Step Size Method by employing the Supercluster, Subtraction and Supersite algorithms (SSS-VSSM).
Links
F. Hess. J. Comp. Chem. 40 (2019) 2664– 2676. 10.1002/jcc.26041